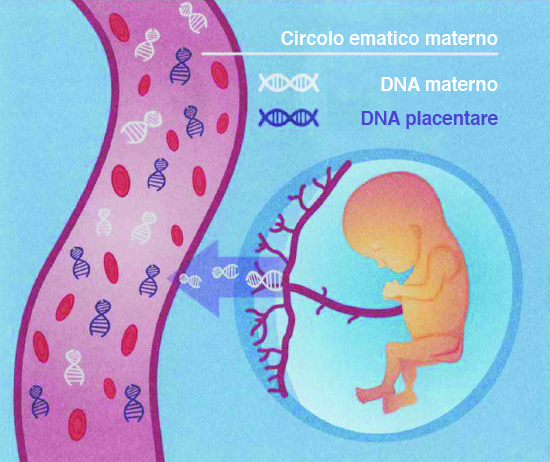




| KARYON® è un test di screening prenatale che analizza le aneuploidie dei cromosomi 21-18-13, dei cromosomi sessuali e consente la determinazione del sesso. |  |
| BIO KARYO® è un test di screening prenatale che analizza le aneuploidie di tutti i cromosomi autosomici, dei cromosomi sessuali e consente la determinazione del sesso. |  |
MICRO KARYON® è un test di screening prenatale che analizza le aneuploidie dei cromosomi 21-18-13, dei cromosomi sessuali, lo studio delle microdelezioni più frequenti e consente la determinazione del sesso. | 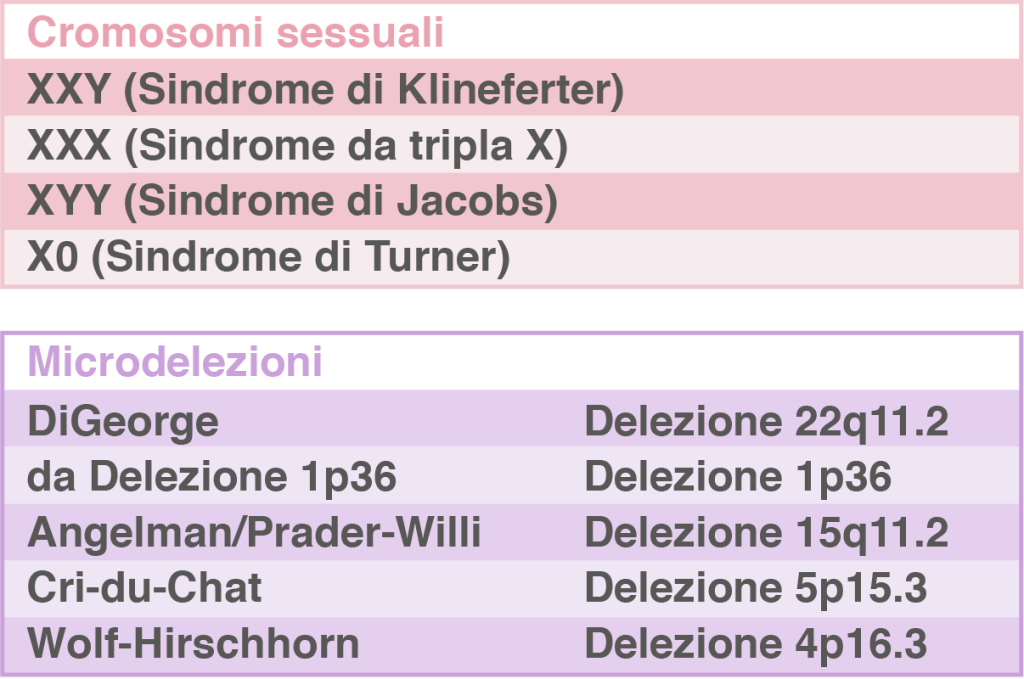 |
| MICRO BIO KARYO® è un test di screening prenatale che analizza le aneuploidie di tutti i cromosomi autosomici, dei cromosomi sessuali, lo studio delle microdelezioni più frequenti e consente la determinazione del sesso. | |